The laboratory mouse (Mus musculus) has been a crucial model organism for studying human biology and diseases for decades. Humans and mice share a highly similar genetic makeup, with approximately 90% of their genomes exhibiting conserved synteny.1 While simpler organisms like yeasts, worms, and flies serve as excellent models for basic biological processes, mice offer a more sophisticated tool for investigating complex physiological systems common among mammals.
Years of research2,3 and advancements in generating mutated mouse strains4 have led to the development of numerous mouse models mimicking various human diseases.5 Even diseases not naturally found in mice, such as cystic fibrosis and Alzheimer’s, can be modeled. The CRISPR-Cas9 technology has revolutionized mouse model creation, enabling efficient genome editing through site-directed DNA endonucleases, directly performed on zygotes, eliminating the need for germline-competent embryonic stem cell lines.6 Mouse models are widely used across diverse fields of biology (Box 1), including neuroscience, behavioral research, physiology, and cancer research. Statistics from the European Committee7 indicate that nearly 11.5 million laboratory animals were used in Europe in 2011, with mice accounting for 61%. A UK governmental report shows 1.16 million mice used in the United Kingdom in 2014, comprising 60% of the 1.93 million experimental procedures completed that year,8 with consistent usage levels since 2005.
Box 1 – Mice as Models for Diseases
Since the early days of mouse research, mice have been engineered to generate models for a variety of human diseases and conditions.163 The Jackson Laboratory has generated more than 5,000 mouse models with different genotypes for almost 1,500 human diseases.5 Their range of application is very broad, including neurological and muscular disorders, genetic illnesses, behavioural and cognitive abilities, response to viruses and cancer research.
Genetic mouse models of neurodegenerative disorders, such as Alzheimer’s disease (Online Mendelian Inheritance in Man (OMIM https://www.omim.org/)) 104300)164 and Parkinson’s disease (OMIM 168600)165, which recapitulate the essential features of each disease, have significantly advanced our understanding of the molecular basis of disease progression. However, their translational impact remains limited, as neurodegenerative human diseases are heterogeneous in both pathological and clinical (or behavioral) domains and the non-hereditary causes (affecting the majority of the cases) are unknown166.
As another example, several mouse models for Down syndrome (also known as trisomy 21, OMIM 190685) have been generated based on the homology of the human chromosome 21 and the mouse chromosomes 10, 16 and 17 167. These models exhibit many of the behavioural, learning and physiological defects associated with the syndrome in humans, and as such have proved useful to test therapies that rescue these alterations168.
As mice can be housed in small and controlled spaces, very manageable behavioural tests have been creatively devised to reproduce major human behavioural patterns. Examples of applications of behavioural tests include studies of anxiety169,170, substance abuse and addiction171, and diet172.
Despite acknowledged discrepancies between the human and murine immune systems173, mouse models exist to also investigate viral infections and limit the ethical and practical costs of primate research. For instance, humanized mice derived from the combination of transplantation of human fetal pluripotent hematopoietic stem cells with surgical engraftment of human fetal thymic tissue (BLT mice) have been used to study many aspects of HIV infection, including prevention, transmission and therapies174. Similarly, human hepatocytes are transplanted into immunodeficient mice to develop humanized chimeric mice, which enable the study of viral replication and cellular changes caused by the human hepatitis viruses175.
Finally, mice have also been widely used for the research of very complex multifactorial conditions, such as autism[176](#R176] and ageing177, where it is crucial to be able to account for one individual factor at a time. Among complex diseases, cancer research is certainly prompting the development of several mouse models to study the relationship between mutations and tumour biology178.
However, current limitations of mouse models are well known [179](#R179]. The use of mice to study the intricacies of human cancer pathogenesis, for example, is limited by many species differences, including cell duplication time, lifespan and cancer susceptibility180, amongst others. Other examples include mouse models of cystic fibrosis (CF; OMIM 219700), a hereditary lung disease caused by a mutation in the gene encoding the membrane protein CFTR. Although these have proven useful to discover ways to correct this defect181, CF mouse models have a limited ability to recapitulate spontaneous lung disease182. Similarly, mouse models for the progressive muscle-wasting disorder Duchenne muscular dystrophy (DMD; OMIM 310200), that is, mdx mice, have been engineered to study potential gene therapies, but a caveat is that they show only minimal clinical symptoms183. Alternative animal models are being investigated to potentiate translational research, and larger mammalian species, such as pigs, ferrets and dogs, are proving beneficial to scale up initial results obtained in mouse models182,183.
It is unsurprising that the mouse is the most commonly used species for scientific purposes. Clinical trials, in particular, rely heavily on non-human organisms, before testing a drug on patients, as proven efficacy in in vivo preclinical studies is essential for a drug to enter further clinical phases. Nonetheless, drugs often fail along the phases of clinical trials; for instance, 40% of the drugs investigated between 2003 and 2011 did not proceed to the second phase of testing, and only 10.4% of drug candidates are likely to get FDA approval9. In cancer research, specifically, the average rate of successful translation from animal models to human clinical trials is less than 8%10, which mimics the difficulties in using mice as xenograft models of cancer11.
The above highlights that although many core biological processes and genetic elements are conserved between human and mouse, other biological features have diverged substantially, leading to phenotypic differences and poorly correlated physiological responses between species. Diverging features can be genomic differences (such as retrotransposition events, gene expansions or gene losses, genomic rearrangements, differences in coding and non-coding sequences) or regulatory differences that affect gene expression and, ultimately, protein levels (such as alternative splicing, enhancer activity, structural elements such as chromatin domains, and post-translational modifications).
With the continuously decreasing cost and technical challenges of high-throughput sequencing technologies has come a growing effort to functionally characterize the human and mouse genomes, to identify what is shared and what has diverged between these two species. To this end, a series of large-scale projects has analysed a vast array of human and mouse samples, with the dual aim of understanding the principles of genomic regulation across different conditions and of comparing them between species. These projects include, but are not limited to, the Genotype-Tissue Expression (GTEx) project12, which aims to establish a resource database and associated tissue bank to study the relationship between genetic variation and gene expression in human tissues, as well as the Roadmap Epigenomics project13 and the Blueprint project14, which aim to build a public resource of human epigenomic data. Other projects that are collecting human and mouse data simultaneously include the FANTOM project15, which focuses mostly on Cap Analysis of Gene Expression (CAGE) profiles of human and mouse tissue and cell lines, and the human and mouse ENCODE projects16,17, the scope of which is to catalogue all functional elements in the human and mouse genomes, respectively.
Characterizing gene expression profiles across multiple samples and species is instrumental to determine to what extent the biology of a given organism can be extrapolated to another. Thus, this Review centres on presenting an overview of the main findings of comparative molecular studies between human and mouse, with a focus on comparative transcriptomics, and how these studies illuminate the cases and conditions under which mouse is a suitable model of human biology. We also discuss the limitation of current approaches, which include incomplete transcriptome characterization, and difficulties in identifying homologous phenotypes and cell types, and how these can be addressed using emerging technologies.
1. Human and Mouse Genomes
The mouse was the second mammalian species to have its genome sequenced in the early 2000s, following humans,18–20 highlighting its importance as a model organism. The most recent genome assemblies (GRC38) include 3.1 Gb and 2.7 Gb for human and mouse, respectively (Table 1), with the murine genome being 12% smaller than the human one. Around 90% of each genome can be partitioned into conserved syntenic regions, and 40% of the nucleotides in human can be aligned to mouse20. The remaining 60% of unalignable nucleotides might be attributed to lineage-specific deletion of repeated elements from the ancestral genome, nucleotide-level insertions and deletions and lineage- specific duplications20.
Table 1. Summary Statistics of Human and Mouse Genomes and Gene Sets.
Annotation counts are retrieved from the Gencode website (http://www.gencodegenes.org/, v25 for human and vM11 for mouse). The number of microRNAs is obtained from miRBase v2150. The number of tRNAs is obtained from GtRNAdb53. The number of protein-coding orthologues is taken from Ensembl Compara24 (v86), while the numbers of orthologous long non-coding RNAs were obtained from different sources23,36–38.
| Human(GRCh38) | Mouse(GRCm38) |
|---|---|
| Genome size (nt) | 3,088,269,832 |
| Unplaced scaffolds (nt) | 11,464,317 |
| Number of chromosomes | 22 + X +Y |
| Chain alignments (nt) | 2,735,135,097 |
| Number of genes | 58,037 |
| Number of transcripts | 198,093 |
| Protein-coding | |
| – genes | 19,950 |
| – 1 to 1 orthologs | 15,893 |
| – transcripts | 80,087 |
| Long non-coding RNAs | |
| – genes | 15,767 |
| – orthologs | 2,720 [36], 1,587 [38], 1,100 [37], 851 [23] |
| – transcripts | 27,692 |
| Pseudogenes | 14,650 |
| Small RNAs | 7,258 |
| – miRNAs [42] | 2,588 |
| – snRNAs | 1,900 |
| – snoRNAs | 944 |
| – tRNAs [46] | 631 |
1.1. Protein-Coding Genes
According to the latest release of GENCODE annotation21 (v25, Ensembl86), which recently started to also curate the mouse genome22 (vM11, Ensembl86), the human genome encodes 58,037 genes, of which approximately one-third are protein-coding (19,950), and which yields 198,093 transcripts. By comparison, the mouse genome encodes 48,709 genes, of which about one half are protein-coding (22,018 genes), and yields 118,925 transcripts overall (Table 1). For both species, the current number of protein-coding genes is about 10,000 genes lower than was estimated from early genome assembly drafts18,20.
The discrepancy in the total number of annotated genes between the two species is unlikely to reflect differences in underlying biology, and can be attributed mostly to the less advanced state of the mouse annotation. The number of protein-coding and long non-coding RNAs (lncRNAs) encoded in the human and mouse genomes is expected to be very similar, and differences in the total genome length do not result from differences in the number of genes, but probably from differences in the lengths of introns and intergenic space20 (Figure 1). Indeed, when including predicted gene models from RNA sequencing (RNA-seq) and CAGE data, the mouse annotation is expanded to a size that is similar to the human annotation23. There is a high degree of gene orthology between human and mouse: 80% of human and 72% of mouse protein-coding genes have a one-to-one orthologous relationship in the automatically derived Ensembl Compara24 (15,893) (Figure 1), a number which is highly similar to the 15,736 orthologous genes derived after extensive curation efforts by the ENCODE consortium25. The remaining 20-30% protein-coding genes are either in one-to-many or many-to-many orthologous relationships, are members of gene families that have undergone species-specific expansions or reductions, or contain species-specific open reading frames (ORFs). These genes might contribute to human disease phenotypes and should therefore be taken into account when engineering mouse models20. For example, the human-specific gene saitohin (STH), which contains a single nucleotide polymorphism (Q7R) that is associated with susceptibility to several neurodegenerative diseases26, has no orthologous gene in mice.
Figure 1. Homology of human and mouse genes and genomic elements. Orthologous genes between human and mouse can be identified based on sequence homology of coding exons.
Orthologous genes tend to have conserved exonic structure and exon lengths, but introns are generally shorter in mouse. There is some degree of conservation of alternative splicing patterns (Box 2), but species specific splicing events exist (green gene). Orthologous genes may have conserved expression profiles between the two species (green) or diverged expression (orange). The bar chart represents expression levels of the genes in different organs. Genes with homologous sequence within the same species are called paralogous. Paralogous genes may originate from gene duplication events and their exonic structure, sequence and expression may diverge with evolutionary time. Promoter sequences (upstream from genes) are less conserved than gene body sequences. Regulatory motifs may differ although regulatory networks may be conserved. Orthologous genomic regions (and elements) can be identified through whole genome alignments (pink). However, some elements cannot be aligned to the other species (different shades of grey), or can map in multiple locations (brown). Finally, some genomic regions can be aligned, but their function may not be conserved (blue).
1.2. Long Non-Coding RNAs
The importance of lncRNAs is increasingly recognized, with a growing number being linked to human diseases annually.27–29 Identifying mouse orthologues of human lncRNAs would greatly aid in understanding their biological roles.
Currently, GENCODE annotates 15,767 lncRNAs in humans and 9,989 in mice.21,22 This difference stems from the less complete mouse genome annotation. LncRNAs are generally expressed at lower levels than protein-coding genes and often in a tissue-specific manner, which complicates their identification and requires additional resources for comprehensive annotation.30,31 Finding orthologous relationships and conservation estimates for lncRNAs is challenging because their sequence is less conserved than that of protein-coding genes30 and not constrained by amino-acid translation. LncRNA orthology is defined by a combination of sequence, functional conservation, and synteny.32 While RNA secondary structure might help identify short non-coding RNAs and their conservation, only a few lncRNAs have distinct structural domains as defined in Rfam.33,34 Therefore, current catalogues of orthologous lncRNAs are incomplete and inaccurate,34 and identifying lncRNA orthology is an active research area.
Several studies have sought to identify novel lncRNAs in mice and other species and their orthologs in humans.23,35–37 While gene sets vary, a consistent estimate is approximately 1,000–2,000 orthologous lncRNAs between humans and mice. Necsulea et al.36 report the highest number of human-mouse orthologous lncRNAs (2,720), based on sequence similarity of both novel and annotated transcripts, whereas Washietl et al.37 identify 1,100 orthologous lncRNAs based on genome-wide chain alignments. Pervouchine et al.23 reported 851 lncRNAs orthologs on the basis of a mixed approach including both genome alignments and sequence homology. A more recent study, which includes the information on syntenic blocks to call the orthology, reports 1,587 human-mouse orthologous lncRNAs[38](#R38]. However, the overlap between these studies is quite low: Pervouchine and colleagues23 reported that only 189 orthologous lncRNAs are in common between their study and that of Necsulea et al.36. In all of these studies, orthologous lncRNAs represent only a small fraction of all annotated lncRNAs in both species, especially when compared to protein-coding genes.
About 5,000 lncRNA transcripts are in antisense orientation with respect to protein-coding genes in both mouse and human,39 and antisense transcription is known to have a role in the regulation of expression of the sense gene in a number of cases40. For example, an antisense transcript of the tumour suppressor gene CDKN1A recruits a regulatory complex that induces trimethylation of Lys27 of histone H3 (H3K27me3) to suppress the sense promoter region40. Although antisense transcription is largely present in both species, the proportion of orthologous sense–antisense pairs relative to all sense–antisense pairs is low (less than 20%, around 1,000 pairs23,39), suggesting low conservation of antisense transcription, and consequently of the corresponding biology.
1.3. Small Non-Coding RNAs
Small non-coding RNAs, including microRNAs (miRNAs), transfer RNAs (tRNAs), small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs), have received less attention in comparative transcriptomics studies compared to protein-coding and long non-coding RNAs. This is partially due to the difficulty in monitoring them, with analyses limited to a few tissues like brain, liver, kidney, heart, and testis.41–44 These small RNAs regulate RNA processing, expression, and translation,45,46 and there is increasing evidence of their involvement in human diseases.29 For instance, altered miRNA expression can lead to diseases ranging from immune-related conditions like multiple sclerosis to neurodegenerative diseases such as Parkinson’s disease47, and cancer48. Therefore, using specific mouse models to understand the mechanisms of small non-coding RNA involvement in diseases will be beneficial.49 For example, obese mice deficient in miR-375 developed severe insulin-deficient diabetes, suggesting that miR-375 is essential for mediating metabolic stress49.
Currently, almost 3,000 and 2,000 miRNAs are annotated in the human and mouse genome, respectively50 (Table 1). However, only a small fraction (300 miRNAs) of them has a defined ortholog in the other species51.
tRNAs possess a unique secondary structure that allows them to recognize mRNA codons and carry a cognate amino acid.52 Due to codon degeneracy, multiple anticodons relate to the same amino acid (tRNAs isoacceptors). Humans and mice share 46 isoacceptors.43 The number of predicted tRNA genes is similar between human and mouse (631 and 471 tRNA genes, respectively, Table 1)53, as is the number of tRNA genes detected in human and mouse liver (223 and 224 tRNA genes, respectively). Although tRNA expression is conserved between the two species at the isotype level (tRNA isoacceptors related to the same amino acid), 34% of mouse tRNA genes cannot be aligned to human homologues, and only 79 tRNA genes are commonly expressed in liver samples43, which suggests a certain degree of divergence in the evolution of tRNA genes.
snRNAs are essential components of the spliceosome, and their expression levels are generally conserved between humans and mice.44 snoRNAs contribute to biochemically modify specific sites of ribosomal RNA, tRNA and snRNA45. Of the 944 and 1,508 annotated human and mouse snoRNA genes21, respectively (Table 1), at least 208 are conserved between the two species44. Of these, 63 snoRNA genes (30%) have distinct expression profiles44, which indicates that the regulation of snoRNA genes has diverged considerably between the two species.
Further studies will certainly improve our understanding of the regulatory role and evolution of these RNA families, and hopefully of their involvement in diseases. Particularly relevant will be the understanding of the conservation between human and mouse of the relationship between the precursor long RNA molecules and the small functional RNA products. This, in particular, would extend the possibility of therapeutic interventions along the entire molecular path involved in the synthesis of small RNA molecules.
2. Conservation of Transcriptomes
Similarities in gene sets between species do not guarantee transcriptomic similarities, as a gene’s expression pattern across tissues and conditions can differ significantly. The development of microarray technologies, and subsequently RNA-seq, has enabled genome-wide surveys of transcriptional activity, sparking interest in understanding the extent to which gene expression and splicing patterns (Box 2) have been globally conserved between humans and mice.
Box 2 – Splicing
Splicing is the mechanism through which exons and introns of genes are processed into mature coding and non-coding transcripts. Different combinations of exonic and intronic sequences can be arranged through alternative splicing to expand the range of processed isoforms from a relatively limited pool of genes.
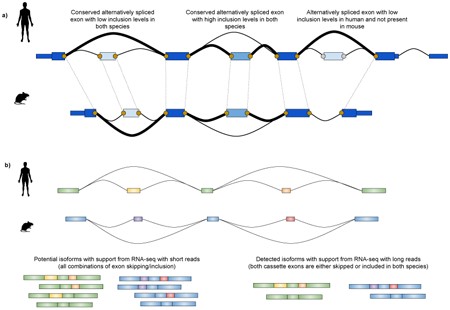
Exon structure and splicing are very similar between humans and mice, in terms of number and order of exons per gene, exon length, precise boundaries and sequence184,185. The exact number of orthologous exons is heavily dependent on the genome assemblies, the annotation status and on the set of analysed genes: the mouse ENCODE consortium has annotated over 150,000 orthologous internal exons17, a noticeable increase compared to the 2,000 exons186 identified in the earliest reports right after publication of the first complete mouse genome draft. Although alternatively spliced exons with low proportion of inclusion tend to be more species-specific184, exon inclusion levels are overall highly correlated between the two species even across very distant sample types23 (see the figure, part a). Indeed, alternative splicing was shown to be less evolutionarily conserved than gene expression in comparative studies including multiple species and organs78,187.
However, comparative analyses of exon inclusion are usually limited to a few hundred conserved exons78,187 and are tied to local splicing events, not considering the whole isoform structure. Determining orthology at the isoform level, for complete gene structures of exons and introns, is particularly challenging, due to the presence of non-coding exons, which have less sequence constraints than coding sequences, and to the redundancy of exonic elements between multiple isoforms of the same gene188. Novel transcriptomic sequencing strategies, for example, synthetic long-read sequencing189 and single-molecule long-read sequencing190, enable detection of full-length transcripts and preserve the relationship between distant exons (see the figure, part b). These techniques, possibly coupled with targeted approaches for lowly abundant loci, will improve the accuracy of isoform detection and might provide new insights on the conservation of isoform usage and of its regulation across species.
2.1. Microarray Studies
Early microarray studies primarily focused on the expression of orthologous protein-coding genes in homologous tissues like brain, heart, muscle, and liver. Assuming the mouse is a good model of human biology, gene expression in homologous organs should be more similar between species than in different organs from the same species.54 In other words, human liver would have an expression profile more similar to mouse liver than to human heart.
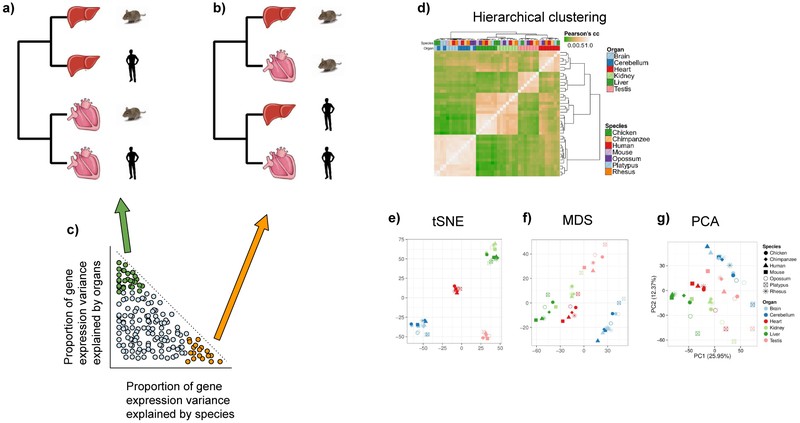
Transcriptome relationships from multiple RNA samples are typically visualized using hierarchical clustering. This approach uses samples as leaves of a dendrogram, built based on a similarity measure between transcriptomes, such as Euclidean distance or correlation coefficient.FIG. 2 Alternative visualization methods include dimensionality reduction techniques like principle component analysis (PCA 55), multidimensional scaling (MDS56), or t-distributed Stochastic Neighbor Embedding (t-SNE57). These techniques project samples onto 2D or 3D spaces where distances reflect overall transcriptome similarity.FIG. 2e-g
Figure 2. Simplified clustering of human and mouse tissue samples and variance decomposition of gene expression.
Samples can be clustered based on their transcriptional profiles. If a human organ (for example, liver or heart) has a more similar gene expression profile to the homologous mouse organ than to another human organ, the clustering is organ-dominated (a). Vice versa, if human organs have more similar gene expression profiles between each other than compared to their homologous mouse organs, the clustering is species-dominated (b). The variation of expression for each gene can be decomposed into the most contributing factors, in this case species and organs (c). Genes are distributed in a continuous way along these proportions of variation. Nonetheless, genes at the extremes of this distribution can be identified as genes with proportionally higher variation across species and lower across organs (orange) and genes with proportionally higher variation across species and lower across organs (green). If only the expression of one or the other set of genes is used for clustering, genes with proportionally higher variation across species or organs lead to a more species-dominated clustering, or organ-dominated clustering, respectively. d∣ Hierarchical clustering based on real gene expression data from different organs across mammals and chicken, performed with the entire set of orthologous genes across species, reveal organ-dominated clustering82. Distances between samples can be visually represented also on a 2-D space through several dimensionality reduction techniques, such tSNE (e, same input as d, perplexity=4, iterations=1000), MDS (f, same input as d, euclidean distance) and PCA82 (g).
These statistical methods heavily depend on input data quality, the amount of variation between and within samples, and value distribution. Proper filtering and normalization are crucial for microarray data, which are subject to technical biases. Studies that emphasize proper normalization report high expression conservation between human and mouse tissues54,[58](#R58], such as brain, muscle, liver, kidney, lung, and spleen, after correcting for array-specific expression differences. In contrast, inaccurate normalization can spuriously exacerbate differences between species59,60.
Whether these results, which support transcriptional conservation between humans and mice but were obtained in a limited number of samples, are generally applicable to all sample types and the entire transcriptome is still debated. For example, while induction and repression of major erythropoiesis transcriptional regulators are conserved between mouse and human, significant transcriptional divergence has been detected at the transcriptome level.61 Many transcriptional differences were also reported at the level of the immune and inflammatory response. These might be explained by cis-regulatory differences. For instance, although the macrophage response to lipopolysaccharide (LPS) is overall conserved between the two species, differential sets of genes are activated and repressed in mouse and human, a transcriptional plasticity that might be conferred by TATA-enriched and CpG island-depleted promoters62. By contrast, intraspecies differences in macrophage transcriptional response to glucocorticoid seem to be associated with gain and losses of glucocorticoid response elements63. In another study, it was shown that mouse transcriptional responses to different inflammatory stresses, including trauma, burns and endotoxemia, correlate poorly with the human ones, even though human transcriptional responses to different inflammatory stresses correlated well with each other64. This finding raised the serious question whether mouse is a good clinical model to study such conditions. This conclusion was challenged by a reanalysis of the same data that was restricted to a smaller set of genes with changes in expression levels that were conserved between human and mouse65. However, it has been noted that this approach introduces a bias in the results, and that the low percentage of genes with conserved changes in expression (12%) may itself be indicative of poor reproducibility of the human response in mice66,67.
2.2. RNA-Seq Studies
RNA-seq technology has enabled more comparative transcriptomics studies with deeper resolution and larger numbers of species, as it does not rely on species-specific spotted microarrays (see68 and69 for reviews). RNA-seq offers greater sensitivity, a broader detection range from low to high expression, and annotation-independent detection of RNA abundances compared to microarray technology.70
The Mouse ENCODE consortium71 has compiled approximately one hundred RNA-seq datasets for various mouse tissues and cell types to serve as a comprehensive reference for future studies.17 The profiled samples included almost 30 tissues from adult mice, as well as brain, nervous system, limbs, and liver from embryos, and mouse cell lines like embryonic stem cells, murine erythroleukemia cells (MEL), and mouse lymphoma cells (CH12). Depending on the sample, they were collected and sequenced at different centres, and at least 2 replicates were sequenced for each sample. As in the case of microarray, clustering of mouse and human gene expression profiles from homologous tissues strongly depended on the normalization method applied17. However, as human data from comparable experimental conditions is not available (the bulk of human ENCODE transcriptome data was obtained in cell lines16 whereas the mouse data were obtained from primary tissues), it is hard to disentangle the gene expression variation attributable to the species from that resulting from other biological factors or technical effects17. Simultaneous analysis of the human and mouse RNA data uncovered a large fraction of orthologous protein-coding genes (about 50%) with fairly constrained expression inde pendent from the sampled cell type in both human and mouse23. Analysis of human and mouse gene expression from a more homogeneous experimental setting, where samples are collected, processed and sequenced similarly at the same centre, however, argued that different conclusions can be drawn depending on which organs are profiled: organs with more distinct signatures of tissue-specific genes, such as brain, testis, heart, liver and kidney showed strong conservation between the two species72–75. By contrast, a study that also included organs expressing fewer tissue-specific genes72, such as fat and stomach, showed that transcriptional patterns are overall different between human and mouse, separating the species more than the organs. This conclusion led to another highly charged debate, suggesting that other factors and biases, such as sequencing site, time of sequencing and the sequencing instrument used, need to be taken into account when undertaking comparative transcriptomics76.
The analysis of additional vertebrate species at different phylogenetic distances to human and mouse, such as macaque, chimpanzee, opossum, platypus and chicken, affirmed the original conclusion that transcriptional patterns are more similar between orthologous organs of different species than between different organs from the same species77,78,79. These studies, however, were again based on organs expressing the largest numbers of organ-specific genes.
Taken together, these studies suggest that the question of whether mouse is overall a suitable model of human biology, based on transcriptome comparisons, is ill-posed. These works implicitly assume an average behaviour for genes, ignoring that each gene has a characteristic pattern of expression variation across species and organs (FIG. 1). This pattern has been recently investigated both between human and mouse17 and across multiple species80 (FIG. 2). In both studies linear models were used to decom pose the variation of gene expression in a set of homologous adult tissues across human and mouse only or across multiple mammals, including human and mouse,