Introduction
Infectious diseases remain a significant cause of morbidity and mortality in children globally, despite advancements in recent decades. Vaccines are a cost-effective solution, and early-life immunizations have reduced deaths worldwide. Recognizing the differences between early life and adult immune systems is critical for developing effective pediatric vaccines and optimizing vaccination schedules. While innate immunity has been a focus, understanding early-life adaptive immunity, specifically adaptive immune memory, is crucial because vaccine-induced protection depends on acquired immunity. This guide provides a detailed overview of T and B cell development, the unique factors regulating early-life adaptive immunity, and discusses how maternal antibodies impact early life adaptive immunity. The aim is to inform the rational design of vaccines and other immune-based interventions to combat pediatric infectious diseases by emphasizing the importance of understanding adaptive immune memory development.
T Cells in Early Life
T Cell Development and the Neonatal T Cell Compartment
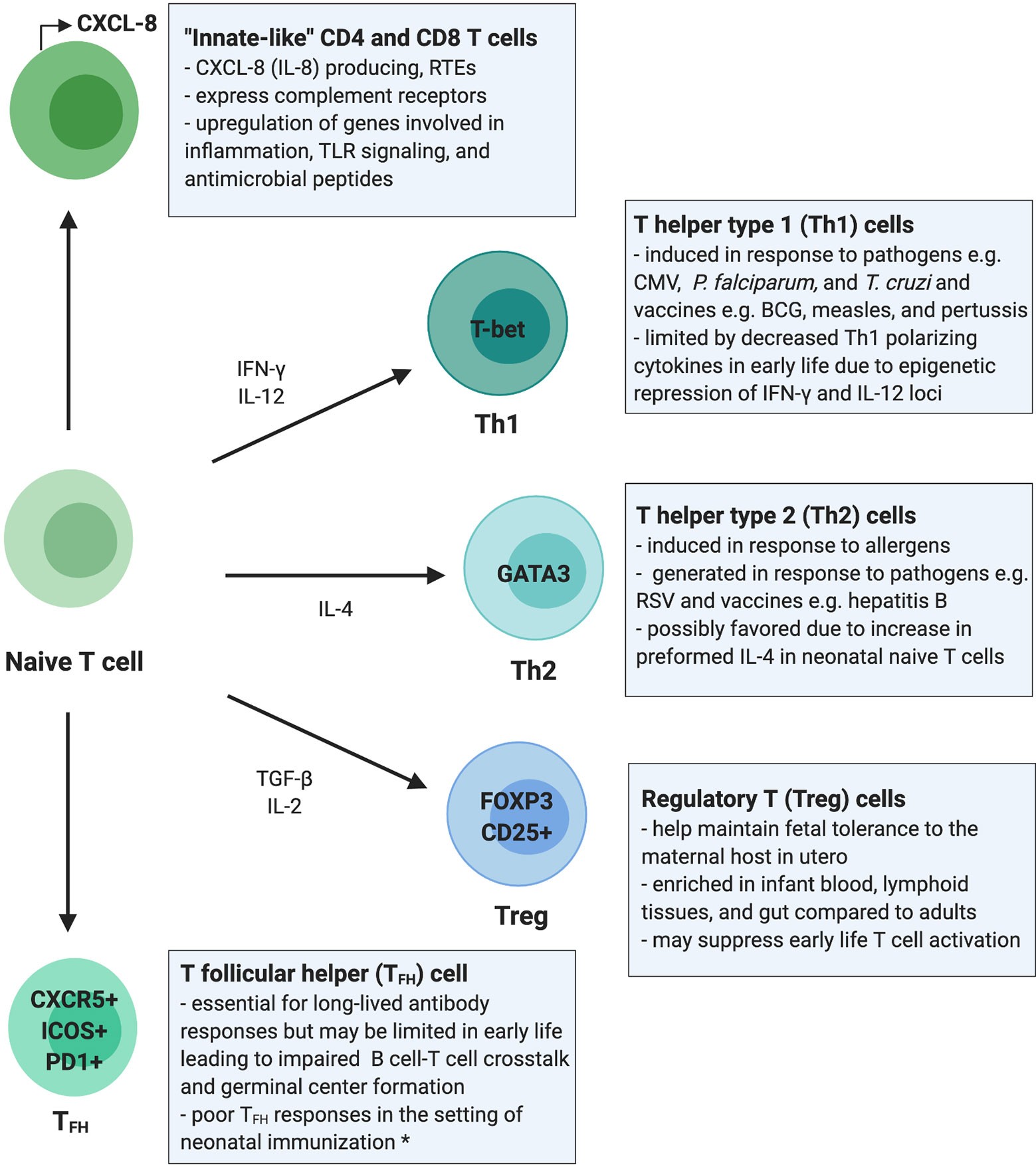
Early Life T Cell Development and Immunophenotypes
The T cell compartment in early life is uniquely positioned to respond to diverse immunological demands, balancing immunotolerance in utero with defense against pathogens. Human T cell development begins in utero, with the thymus producing T cells from 12–14 weeks of gestation, and the T cell receptor (TCR) repertoire is diversified by 26 weeks. Infant and adult T cell compartments remain phenotypically distinct for at least the first 2 years of life. Infants have high inter-individual variability in CD4+ and CD8+ subsets, yet the neonatal T cell compartment is dominated by naive T cells, the majority being recent thymic emigrants (RTEs). RTEs are biased towards innate-like immune signaling and preferentially differentiate into induced regulatory T cells (iTregs), limiting their role in adaptive T cell responses. Despite the predominance of naïve T cells, TEM are present in cord blood even in the absence of intrauterine infection, suggesting that T cell memory is generated during normal fetal development.
Innate-Like Functions of T Cells in Early Life
Adult and infant T cells have distinct functional responses. Neonatal peripheral blood contains fewer IFN-γ and IL-17A-producing T cells but more CXCL-8-producing T cells compared to adults. CXCL-8 producing T cells can co-express complement receptors (CR), which also function in innate immune cell signaling. Over the first 3 months of life, CXCL-8 plasma concentrations decrease whereas IL-17A concentrations increase, reflecting a shift from innate-like to canonical adaptive cytokine production. These differences highlight the importance of CXCL-8 and suggest that neonatal T cells respond preferentially with innate rather than adaptive responses favored in adulthood. This skewing of the early-life T cell compartment towards innate-like responses may need to be overcome to elicit durable adaptive T cell responses in infancy.
Figure 1: This overview of the infant T cell compartment summarizes evidence from human studies. Infants rely on “innate-like” CD4+ and CD8+ T cells which are more likely to be RTEs and to signal through innate immune pathways such as complement receptor and TLR signaling, promote inflammation, and respond with antimicrobial peptides rather than classic antigen-specific adaptive immune responses.
Mechanisms Regulating T Cells in Early Life
Cell-intrinsic and cell-extrinsic mechanisms regulating T cell responses differ between early life and adulthood. Fetal and neonatal T cells have a transcriptional landscape that favors tolerogenic and innate-like cytokine production over proinflammatory T cell responses. Neonatal T cells are biased towards innate immune signaling and immunosuppression. Transcriptional differences between naïve CD8+ T cells from cord and adult blood support the hypothesis that T cells in early life harbor a gene expression program favoring innate over adaptive T cell functions. Neonatal T cells also have a distinct epigenetic landscape compared to adult T cells. In fetal CD4+ T cells, IL-2 and IFN-γ production is blunted by expression of the transcription factor Helios. Studies are needed to understand the transcriptional and epigenetic mechanisms governing T cells postnatally, potentially revealing novel therapeutic targets for modulating infant T cells to promote long-lasting adaptive responses.
Immunotolerance and Regulatory T Cells (Tregs) in Early Life
A strong bias towards Treg development in early life tempers adaptive immune responses. Tregs maintain fetal immunotolerance in utero, hampering proinflammatory T cell activity and promoting tolerance to the maternal host. Tregs are enriched in fetal lymphoid tissues and suppress non-Treg T cell proliferation, activation, and IFN-γ production. Both fetal, and to a lesser extent, neonatal CD4+ T cells are poised to differentiate into Tregs upon TCR engagement. Recent work demonstrates that Tregs are a highly enriched and compartmentalized T cell subset in early postnatal life, accounting for a higher percentage of total CD4+ T cells in pediatric blood, lung, intestine, and lymphoid tissues compared to adult donor tissues.
T Helper (Th) Cell Responses in Early Life
CD4+ T cells are believed to be biased towards T helper type 2 (Th2) differentiation in early life. Naïve CD4+ T cells can differentiate into Th subsets including Th1, Th2, and Th17 cells, which produce IFN-γ, IL-4/IL-5/IL-13, and IL-17/IL-22, respectively. Th responses in early life appear to be highly stimulus dependent. Recently, a population of naïve CD4+ T cells has been identified in cord blood and infant adenoids that highly express IL-4 when stimulated under multiple conditions. T cells isolated from cord blood produce Th2 type cytokines when stimulated by environmental allergens ex vivo. Th responses in early life are largely dictated by the stimulation and cytokine milieu.
T Cell Responses to Pathogens and Vaccines in Early Life
Early Life T Cell Responses to Pathogens
Studies of fetal and neonatal immune responses to infections have advanced understanding of T cell functional capabilities in early life. Fetuses mount pathogen-specific CD8+ and CD4+ T cell responses against human immunodeficiency virus (HIV), cytomegalovirus (CMV), Trypanosoma cruzi, and Plasmodium falciparum. Infants exposed to P. falciparum in utero produce antigen-specific CD4+ and CD8+ T cell responses that undergo memory differentiation. Infectious exposures can impact the developing T cell compartment broadly in addition to eliciting pathogen-specific T cell responses. Congenital CMV infection causes widespread immune activation and differentiation of the developing T cell compartment. Infant T cell responses are limited compared to adults. Infants have impaired CD4+ T cell responses to P. vivax, HIV, and CMV, which may contribute to poor pathogen control.
Early Life T Cell Responses to Vaccines
Infant T cell responses to vaccines demonstrate that the T cell compartment can mount pathogen-specific adaptive responses in early life, though diminished compared to adulthood. Infants generate T cell responses following measles vaccination, yet immunization before 6 months of age produces less IFN-γ than adults. Infants receiving the hepatitis B (HepB) vaccine have impaired T cell cytokine production and differentiation compared adults. Infants receiving the Bacillus Calmette–Guérin (BCG) vaccine against Mycobacterium tuberculosis have more robust Th1 and pathogen-specific CD8+ T cell responses than adults. Bordetella pertussis vaccination also induces a potent Th1 response in young infants and can enhance overall T cell activation. Therefore, targeted intervention strategies that account for the distinct nature of the neonatal T cell compartment should be employed to effectively engage T cells in adaptive responses.
B Cells and Antibody Responses in Early Life
B Cell Development and the Neonatal B Cell Compartment
Development of Antibody Responses and B Cell Receptor (BCR) Diversity in Early Life
Immunoglobulins (Ig) encoded by the B cell receptor (BCR) genes, are the key mediators of adaptive humoral immunity. While fetal IgM production begins in utero and increases substantially postnatally, endogenous IgG and IgA production, which requires B cell class-switching, remains limited until 6 months of age. BCR combinatorial diversity is achieved when different genes segments in V(D)J loci are recombined to form the V region and junctional diversity. BCR combinatorial diversity begins around 12 weeks of gestation. At birth, infants have low levels of SHM, but over time, SHM rates increase. The lower rates of SHM in infants can lead to reduced antibody binding to the antigen.
T Cell-Dependent and T Cell-Independent Humoral Responses to Antigens in Early Life
Overall, early life B cell responses to T cell-dependent (TD) and T cell-independent (TI) antigens are weaker than in adulthood except for a few pathogens and vaccines. Multiple factors contribute to the diminished antibody response in infancy, including cell-intrinsic and cell-extrinsic factors. The gene expression profile of neonatal B cells is distinct from adult B cells, which limits the activation signals neonatal B cells receive from CD4+ T cells upon exposure to TD antigens. The follicular dendritic cell (FDC) network, germinal centers, and the bone marrow are involved in eliciting a potent and durable antibody response. Another mechanism that may limit early life humoral responses could be the lower expression levels of CD73 in neonatal naïve B cells.
Humans do not develop humoral responses to TI polysaccharides until 1–2 years of age. Neonatal hyporesponsiveness to TI antigens may be due to immaturity of marginal zone B cells and lower numbers of CD27+ memory B cells in early life. Lower B cell expression of the C3 complement receptor 2 (CR2 or CD21) and decreased levels of C3 complement may further impair humoral responses to TI antigens in early life. To overcome this limitation of the early life immune system, pediatric vaccines against encapsulated pathogens are conjugated to proteins to recruit T cell help and improve humoral vaccine responses. Therefore, factors limiting humoral responses to both TD and TI antigens in early life and the nature of the antigen must be considered when designing pediatric vaccines.
Figure 2: Summary of the B cell compartment and antibody responses in early life. Dampened germinal center reaction is generally observed in infants, and differentiation of activated B cells is skewed toward short-lived memory B cells over plasma cells. Bone marrow in early life is unable to provide an optimal niche for the differentiation of long-lasting plasma cells. Transplacental maternal antibodies influence the neonatal B cell-mediated response by the illustrated mechanisms.
Unique Factors Influencing Early Life Humoral Responses
Early life antibody responses can be substantially shaped by pathogens, maternal antibodies, and microbial colonization. Factors unique to early life that shape humoral responses include maternally transferred antibodies and colonization of the newborn gut microbiota. Neonates rely heavily on passive maternal antibody transfer for protection against infections. Transplacental IgG transfer, mediated by the neonatal Fc receptor (FcRn), begins during the second trimester and increases throughout gestation. Maternal IgG persists in infant circulation for months postnatally, and the importance of maternal antibodies in protecting neonates is underscored by diseases in which transplacental antibody transfer is disrupted. Passive maternal antibody transfer of mostly secretory IgA (sIgA) and some IgG postnatally through breastmilk also provides protection of the neonate particularly at mucosal surfaces. The presence of maternal antibodies can also dampen B cell responses to vaccines in human neonates.
The newborn gut microbiome is primarily colonized shortly after birth, yet a stable, adult-like community is not established until around 3 years of age. The microbiome can substantially modulate the host antibody repertoire and responses to infections and vaccines in early life. A bias towards HIV envelope (i.e., gp41) specific antibodies that cross react with commensal bacteria has been observed in HIV infected and uninfected adults, but gp41 dominant antibody responses are not observed during pediatric HIV infection. This highlights how immune imprinting by the microbiome can modulate antibody specificities and that targeting the microbiome could be a novel strategy for engineering humoral responses to vaccination in early life.
Antibody Responses to Pathogens and Vaccines in Early Life
Early Life Humoral Responses to Pathogens
Infants and young children can mount robust antibody responses to some pathogens. Infants generate antibodies that neutralize diverse HIV strains by targeting conserved epitopes (known as broadly neutralizing antibodies, bnAbs). High levels of SHM seem to be less important in pediatric bnAbs as two bnAbs, BF520.1 and AIIMS-P01, isolated from HIV-infected children demonstrate neutralizing potency comparable to adult bnAbs but have much lower SHM rates (6.6 and 7% versus 15.8 to 23.1%). Potently neutralizing antibodies with limited rates of SHM have been observed in RSV infections in early life as well.
Humoral Responses to Vaccines in Early Life
Infant humoral responses to vaccination are often attenuated compared to adulthood due to immaturity, fewer co-stimulatory receptors, lower SHM rates, limited class-switching, and maternal antibody interference. An ideal neonatal vaccine should induce a robust and durable immune response with a single dose at birth, thereby minimizing vulnerability to infections. Newborns can mount adult-like antibody responses following HepB vaccination. Children immunized with an oil-in-water emulsion adjuvant (MF59) developed higher antibody titers than adults immunized with the same vaccine, yet no difference was observed between adults and infants when immunized with an Alum-adjuvanted vaccine.
Leveraging Knowledge of Early Life Adaptive Immunity
Vaccination Strategies to Engage Infant Adaptive Immunity
Given the particularities of early life adaptive immunity, vaccination strategies must be specifically tailored to this stage of immune development. Early life T cell responses are limited due to differences favoring innate and effector over adaptive memory responses, immunotolerance, and an enrichment of RTEs and Tregs. The ability to generate long-lived plasma B cells and antibody responses in early life is limited due to immaturity, maternal antibody interference, poor germinal center formation, and reduced T and B cell crosstalk.
A promising approach to improve adaptive immune responses to vaccines in early life is through the informed choice of vaccine adjuvants and novel adjuvant combinations, which can stimulate both adaptive immune cells and recruit innate antigen-presenting cells. For instance, the vaccine adjuvant MF59 elicited robust antigen-specific T cell responses and prevented disease acquisition in infants vaccinated against influenza. Adjuvants combining Alum with TLR4 or TLR9 were able to induce TFH responses in neonatal mice. A TLR7/8 agonist adjuvant 3M-052 overcame the hyporesponsiveness to the pneumococcal conjugate vaccine at birth.
Vaccination Strategies Using Passive Maternal Antibody Transfer
Another way to overcome the challenges to generating persistent humoral responses in neonates is to leverage passive maternal antibody transfer. Vaccine-elicited IgG transferred transplacentally and sIgA transferred via breast milk can protect neonates while adaptive immunity matures. Maternal vaccination against influenza and pertussis during pregnancy has been shown to be effective in protecting infants. Considerations of maternal antibody interference with infant B cell development and antibody responses must be considered when integrating maternal vaccination strategies into the pediatric vaccine schedule.
Concluding Remarks
The neonatal immune system is capable of generating antigen-specific T and B cell responses, even though early life adaptive immune responses to pathogens and vaccinations are limited. The immunologic milieu, immune stimulus, and immune cross-talk from maternal antibodies and microbial colonization are key regulators of infant adaptive immune responses, which must be considered when developing immune-based interventions for early life. The informed use adjuvants and optimization of vaccine schedules will be essential to harness infant adaptive immunity through vaccination. Immunization strategies targeting mother-infant dyads and passive maternal antibody transfer are a promising strategy for protecting neonates during this vulnerable period of immune development.
References
(References from the original article, formatted according to a standard citation style).