CRISPR/Cas systems are adaptive immune systems found in bacteria and archaea, providing defense against invading viruses and plasmids through RNA-guided silencing. This study unveils the mechanism of a Type II CRISPR/Cas system, demonstrating that a mature crRNA base-paired to a trans-activating tracrRNA forms a unique two-RNA structure. This structure directs the CRISPR-associated protein Cas9 to induce double-stranded (ds) breaks in target DNA. Specifically, the Cas9 HNH nuclease domain cleaves the complementary strand at sites complementary to the crRNA-guide sequence, while the Cas9 RuvC-like domain cleaves the non-complementary strand. This groundbreaking research also shows that the dual-tracrRNA:crRNA can be engineered into a single RNA chimera, maintaining its ability to direct sequence-specific Cas9 dsDNA cleavage. This discovery unveils a novel family of endonucleases leveraging dual-RNAs for targeted DNA cleavage, offering immense potential for RNA-programmable genome editing.
The Core Mechanism: How Cas9 Achieves Targeted DNA Cleavage
Bacteria and archaea have developed RNA-mediated adaptive defense mechanisms known as CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas (CRISPR-associated) systems, protecting them from viruses and plasmids(1–3). These systems utilize small RNAs for specific detection and silencing of foreign nucleic acids. CRISPR/Cas systems consist of cas genes organized in operons and a CRISPR array comprising unique genome-targeting sequences (spacers) interspersed with identical repeats (1–3). CRISPR/Cas-mediated immunity involves three phases. During the adaptive phase, bacteria and archaea integrate short fragments of foreign sequences (protospacers) into the host chromosome at the CRISPR array’s proximal end in response to viral and plasmid challenges (1–3). The expression and interference phases involve transcription of the repeat-spacer element into precursor CRISPR RNA (pre-crRNA) molecules, followed by enzymatic cleavage to produce short crRNAs. These crRNAs can then base-pair with complementary protospacer sequences of invading viral or plasmid targets (4–11). The Cas proteins, in complex with the crRNAs, recognize and silence foreign sequences based on crRNA target recognition (10, 12–20).
CRISPR/Cas systems are classified into three types (21–23). Type I and III systems share similar features: specialized Cas endonucleases process pre-crRNAs, and mature crRNAs assemble into large multi-Cas protein complexes capable of recognizing and cleaving nucleic acids complementary to the crRNA. Type II systems, however, employ a different pre-crRNA processing mechanism. Here, a trans-activating crRNA (tracrRNA), complementary to the repeat sequences in pre-crRNA, initiates processing by the double-stranded RNA-specific ribonuclease RNase III in the presence of the Cas9 protein (formerly Csn1) (4, 24) (fig. S1). Cas9 is believed to be the sole protein responsible for crRNA-guided silencing of foreign DNA (25–27).
This study reveals that Cas9 proteins in Type II systems constitute a family of enzymes requiring a base-paired structure between the activating tracrRNA and the targeting crRNA to cleave target double-stranded (ds) DNA. Site-specific cleavage depends on both base-pairing complementarity between the crRNA and the target protospacer DNA, and a protospacer adjacent motif (PAM) located next to the complementary region in the target DNA. The research further demonstrates that the Cas9 endonuclease family can be programmed with single RNA molecules to cleave specific DNA sites, raising the exciting possibility of developing a simple and versatile RNA-directed system for generating dsDNA breaks (DSBs) for genome targeting and editing.
Cas9: A Dual-RNA Guided DNA Endonuclease Unveiled
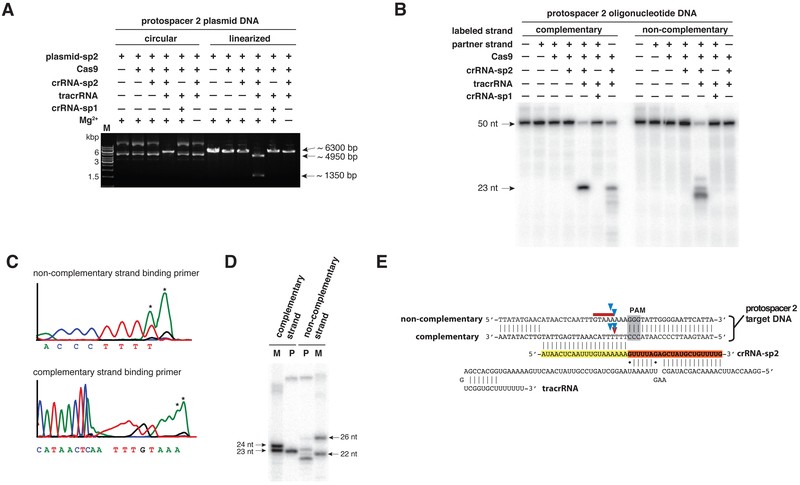
Cas9, the defining protein of Type II systems, was hypothesized to be involved in both crRNA maturation and crRNA-guided DNA interference (fig. S1) (4, 25–27). While Cas9’s role in crRNA maturation was established (4), its direct participation in target DNA destruction remained unexplored. To investigate Cas9’s potential for target DNA cleavage, the researchers used an overexpression system to purify Cas9 protein from Streptococcus pyogenes (fig. S2) and assessed its ability to cleave plasmid DNA or oligonucleotide duplexes containing a protospacer sequence complementary to a mature crRNA, along with a functional PAM. Mature crRNA alone failed to direct Cas9-catalyzed plasmid DNA cleavage (Fig. 1A and fig. S3A). However, the addition of tracrRNA, which can base pair with the crRNA repeat sequence and is vital for crRNA maturation, activated Cas9 to cleave plasmid DNA (Fig. 1A and fig. S3A). This cleavage reaction required magnesium and a crRNA sequence complementary to the DNA. A crRNA capable of tracrRNA base-pairing but containing a non-cognate target DNA-binding sequence did not support Cas9-catalyzed plasmid cleavage (Fig. 1A, fig. S3A, compare crRNA-sp2 to crRNA-sp1 and fig. S4A). Similar results were observed with a short linear dsDNA substrate (Fig. 1B and fig. S3B, C). Thus, the trans-activating tracrRNA is a small non-coding RNA with two key functions: initiating pre-crRNA processing by RNase III (4) and subsequently activating crRNA-guided DNA cleavage by Cas9.
Fig. 1. Cas9 Requires Two RNA Molecules for DNA Endonuclease Activity.
Dual-RNA-guided Cas9 cleaves both plasmid and short linear dsDNA with site-specificity (Fig. 1C-E and fig. S5A, B). Plasmid DNA cleavage produced blunt ends three base pairs upstream of the PAM sequence (Fig. 1C, E and fig. S5A, C) (26). In short dsDNA duplexes, the DNA strand complementary to the crRNA’s target-binding sequence (the complementary strand) is cleaved three base pairs upstream of the PAM (Fig. 1D, E and fig. S5B, C). The non-complementary DNA strand is cleaved at one or more sites within 3 to 8 base pairs upstream of the PAM. Further analysis revealed that the non-complementary strand undergoes initial endonucleolytic cleavage followed by trimming by a 3’-5’ exonuclease activity (fig. S4B). Under single turnover conditions, Cas9 cleavage rates ranged from 0.3 to 1 min−1, comparable to restriction endonucleases (fig. S6A). Incubation of wild-type Cas9-tracrRNA:crRNA complex with a 5-fold molar excess of substrate DNA indicated that the dual-RNA-guided Cas9 is a multiple-turnover enzyme (fig. S6B). Unlike the CRISPR Type I Cascade complex (20), Cas9 cleaves both linearized and supercoiled plasmids (Fig. 1A, 2A). Therefore, an invading plasmid can be cleaved multiple times by Cas9 proteins programmed with different crRNAs.
Fig. 2. Dual Nuclease Domains in Cas9 Cleave Opposite DNA Strands.
(A) Top: Illustration of Cas9 domain structure highlighting mutation positions. Bottom: Endonuclease activity assays of wild-type and nuclease mutant Cas9 proteins with tracrRNA:crRNA-sp2, as in Fig. 1A. (B) Activity testing of wild-type Cas9 and nuclease domain mutants with tracrRNA and crRNA-sp2, as in Fig. 1B.
Unraveling Cas9’s Nuclease Domains: A Strand-Specific Cleavage Mechanism
Cas9 contains domains homologous to both HNH and RuvC endonucleases (Fig. 2A, fig. S7) (21–23, 27, 28). Cas9 variants with inactivating point mutations in the catalytic residues of either the HNH or RuvC-like domains were designed and purified (Fig. 2A and fig. S7) (23, 27). Incubation of these variant Cas9 proteins with native plasmid DNA showed that dual-RNA-guided mutant Cas9 proteins produced nicked open circular plasmids, while the wild-type Cas9 protein-tracrRNA:crRNA complex produced a linear DNA product (Fig. 1A, 2A and fig. S3A, S8A). This indicates that the Cas9 HNH and RuvC-like domains each cleave one plasmid DNA strand. To determine which strand of the target DNA is cleaved by each Cas9 catalytic domain, the mutant Cas9-tracrRNA:crRNA complexes were incubated with short dsDNA substrates where either the complementary or non-complementary strand was radiolabeled at its 5’ end. The resulting cleavage products indicated that the Cas9 HNH domain cleaves the complementary DNA strand, while the Cas9 RuvC-like domain cleaves the non-complementary DNA strand (Fig. 2B and fig. S8B).
Deciphering Dual-RNA Requirements: Target DNA Binding and Cleavage Activation
tracrRNA might be necessary for target DNA binding and/or for stimulating Cas9’s nuclease activity after target recognition. To address these possibilities, electrophoretic mobility shift assays were used to monitor target DNA binding by catalytically inactive Cas9 in the presence or absence of crRNA and/or tracrRNA. TracrRNA significantly enhanced target DNA binding by Cas9, while little specific DNA binding was observed with Cas9 alone or Cas9-crRNA (fig. S9). This indicates that tracrRNA is required for target DNA recognition, possibly by properly orienting the crRNA for interaction with the complementary strand of target DNA. The predicted tracrRNA:crRNA secondary structure involves base-pairing between the 3’-terminal 22-nucleotides of the crRNA and a segment near the 5’ end of the mature tracrRNA (Fig. 1E). This interaction creates a structure in which the 5’-terminal 20 nucleotides of the crRNA, which vary in sequence in different crRNAs, are available for target DNA binding. The bulk of the tracrRNA downstream of the crRNA base-pairing region is free to form additional RNA structure(s) and/or interact with Cas9 or the target DNA site. To determine if the entire tracrRNA length is needed for site-specific Cas9-catalyzed DNA cleavage, Cas9-tracrRNA:crRNA complexes were reconstituted using full-length mature (42-nt) crRNA and various truncated tracrRNA forms lacking sequences at their 5’ or 3’ ends. These complexes were tested for cleavage using short target dsDNA. A substantially truncated version of the tracrRNA retaining nucleotides 23-48 of the native sequence was capable of supporting robust dual-RNA-guided Cas9-catalyzed DNA cleavage (Fig. 3A, C and fig. S10A, B). CrRNA truncation from either end showed that Cas9-catalyzed cleavage in the presence of tracrRNA could be triggered with crRNAs missing the 3’-terminal 10 nucleotides (Fig. 3B, C). Conversely, a 10-nucleotide deletion from the 5’ end of crRNA abolished DNA cleavage by Cas9 (Fig. 3B). The study also analyzed Cas9 orthologs from various bacterial species for their ability to support S. pyogenes tracrRNA:crRNA-guided DNA cleavage. Distantly related orthologs, unlike closely related S. pyogenes Cas9 orthologs, were not functional in the cleavage reaction (fig. S11). Similarly, S. pyogenes Cas9 guided by tracrRNA:crRNA pairs originating from more distant systems failed to cleave DNA efficiently (fig. S11). The species-specificity of dual-RNA-guided cleavage suggests co-evolution of Cas9, tracrRNA, and the crRNA repeat, along with the existence of an unknown structure and/or sequence in the dual-RNA crucial for the formation of the ternary complex with specific Cas9 orthologs.
Fig. 3. Minimal tracrRNA Activating Domain and crRNA Seed Sequence Guide Cas9 Cleavage.
To investigate the protospacer sequence requirements for Type II CRISPR/Cas immunity in bacterial cells, protospacer-containing plasmid DNAs with single-nucleotide mutations were analyzed for maintenance following transformation in S. pyogenes and for in vitro cleavage by Cas9. Mutations near the PAM and Cas9 cleavage sites were not tolerated in vivo and reduced plasmid cleavage efficiency in vitro, contrasting with point mutations introduced at the 5’ end of the protospacer (Fig. 3D). These results align with previous reports of protospacer escape mutants selected in the Type II CRISPR system from S. thermophilus in vivo (27, 29). Furthermore, plasmid maintenance and cleavage results suggest a “seed” region at the protospacer sequence’s 3’ end, critical for interaction with crRNA and subsequent Cas9 cleavage. Supporting this, Cas9 enhanced complementary DNA strand hybridization to the crRNA, strongest in the 3’-terminal region of the crRNA targeting sequence (fig. S12). Consistent with this, at least 13 contiguous base pairs between the crRNA and the target DNA site proximal to the PAM are required for efficient target cleavage, while up to six contiguous mismatches in the protospacer’s 5’-terminal region are tolerated (Fig. 3E). These findings resemble the seed sequence requirements for target nucleic acid recognition in Argonaute proteins (30, 31) and the Cascade and Csy CRISPR complexes (13, 14).
The PAM Motif: Licensing R-Loop Formation for DNA Cleavage
In several CRISPR/Cas systems, distinguishing between self and non-self involves a short sequence motif conserved in the foreign genome, known as the PAM (27, 29, 32–34). PAM motifs are a few base pairs long, with varying precise sequences and positions depending on the CRISPR/Cas system type (32). In the S. pyogenes Type II system, the PAM follows an NGG consensus sequence, containing two G:C base pairs one base pair downstream of the crRNA binding sequence within the target DNA (4). Transformation assays showed that the GG motif is essential for protospacer plasmid DNA elimination by CRISPR/Cas in bacterial cells (fig. S13A), consistent with previous observations in S. thermophilus (27). The motif is also essential for in vitro protospacer plasmid cleavage by tracrRNA:crRNA-guided Cas9 (fig. S13B). To determine the PAM’s role in target DNA cleavage by the Cas9-tracrRNA:crRNA complex, a series of dsDNA duplexes with mutations in the PAM sequence on the complementary, non-complementary, or both strands were tested (Fig. 4A). Cleavage assays using these substrates showed that Cas9-catalyzed DNA cleavage was particularly sensitive to PAM sequence mutations on the DNA’s non-complementary strand, unlike complementary strand PAM recognition by Type I CRISPR/Cas systems (20, 34). Mutations of the PAM motif did not affect cleavage of target single-stranded DNAs. This suggests that the PAM motif is required only in the context of target dsDNA, possibly for duplex unwinding, strand invasion, and R-loop structure formation. Using a different crRNA-target DNA pair (crRNA-sp4 and protospacer 4 DNA), selected due to the presence of a canonical PAM absent in the protospacer 2 target DNA, it was found that both G nucleotides of the PAM were required for efficient Cas9-catalyzed DNA cleavage (Fig. 4B and fig. S13C). Native gel mobility shift assays analyzed the binding affinities of the Cas9-tracrRNA:crRNA complex for target DNA sequences to determine whether the PAM directly recruits the complex to the correct target DNA site (Fig. 4C). Mutation of either G in the PAM sequence substantially reduced the affinity of Cas9-tracrRNA:crRNA for the target DNA. This argues for specific recognition of the PAM sequence by Cas9 as a prerequisite for target DNA binding and potentially strand separation to enable strand invasion and R-loop formation, analogous to CasA/Cse1 recognition of the PAM sequence in a Type I CRISPR/Cas system (34).
Fig. 4. PAM Sequence Critical for Target DNA Cleavage by Cas9 Complex.
Simplifying Genome Editing: Cas9 Programmed with a Single Chimeric RNA
The likely secondary structure of the tracrRNA:crRNA duplex (Fig. 1E, 3C) suggested that the features required for site-specific Cas9-catalyzed DNA cleavage could be captured in a single chimeric RNA. While the tracrRNA:crRNA target selection mechanism works efficiently in nature, a single RNA-guided Cas9 offers potential for programmed DNA cleavage and genome editing (Fig. 5A). Two versions of a chimeric RNA were designed, featuring a target recognition sequence at the 5’ end followed by a hairpin structure retaining the base-pairing interactions between the tracrRNA and crRNA (Fig. 5B). This single transcript effectively fuses the 3’ end of crRNA to the 5’ end of tracrRNA, mimicking the dual-RNA structure needed to guide site-specific DNA cleavage by Cas9. Plasmid DNA cleavage assays showed that the longer chimeric RNA guided Cas9-catalyzed DNA cleavage similarly to the truncated tracrRNA:crRNA duplex (Fig. 5B and fig. S14A). The shorter chimeric RNA was less efficient, confirming that nucleotides 5-12 positions beyond the tracrRNA:crRNA base-pairing interaction are important for efficient Cas9 binding and/or target recognition. Similar results were seen in cleavage assays using short dsDNA, further indicating that the cleavage site in target DNA is identical to that observed using the dual tracrRNA:crRNA guide (Fig. 5C and fig. S14B). Finally, to test whether chimeric RNA design is universally applicable, five different chimeric guide RNAs were engineered to target a portion of the green-fluorescent protein (GFP) gene (fig. S15A-C), and their efficacy against a plasmid carrying the GFP coding sequence in vitro was assessed. In all five cases, Cas9 programmed with these chimeric RNAs efficiently cleaved the plasmid at the correct target site (Fig. 5D, fig. S15D), indicating that rational design of chimeric RNAs is robust and can enable targeting of any DNA sequence of interest with few constraints beyond the presence of a GG dinucleotide adjacent to the targeted sequence.
Fig. 5. Single Engineered RNA Guides Cas9 with Combined tracrRNA and crRNA Functionality.
Conclusion: A Powerful Tool for Programmable Genome Editing
This study has revealed a DNA interference mechanism involving a dual-RNA structure that guides a Cas9 endonuclease to introduce site-specific double-stranded breaks in target DNA. The tracrRNA:crRNA-guided Cas9 protein uses distinct endonuclease domains, HNH and RuvC-like, to cleave the two DNA strands. Cas9 target recognition requires both a seed sequence in the crRNA and a GG dinucleotide-containing PAM sequence next to the crRNA-binding region in the target DNA. The research also demonstrates that the Cas9 endonuclease can be programmed with a single guide RNA engineered as a single transcript to target and cleave any dsDNA sequence of interest. The system is efficient, versatile, and programmable by simply changing the DNA target-binding sequence in the guide chimeric RNA. Existing artificial enzymes like Zinc-Finger Nucleases (ZFNs) and Transcription-Activator Like Effector Nucleases (TALENs) are widely used for genome manipulation (35–38). This RNA-programmed Cas9 methodology offers an alternative approach with considerable potential for gene targeting and genome editing applications.
Supplementary Material
Supplement
NIHMS995853-supplement-Supplement.pdf (6.2MB, pdf)
Acknowledgements
We thank Kaihong Zhou, Alison Marie Smith, Rachel Haurwitz and Sam Sternberg for excellent technical assistance, and members of the Doudna and Charpentier laboratories and Jamie Cate for comments on the manuscript. We thank Barbara Meyer and Te-Wen Lo (UC Berkeley/HHMI) for providing the GFP plasmid. This work was funded by the Howard Hughes Medical Institute (M.J. and J.A.D.), the Austrian Science Fund (W1207-B09, K.C. and E.C.), the University of Vienna (K.C.), the Swedish Research Council (#K2010-57X-21436-01-3 and #621-2011-5752-LiMS, E.C.), the Kempe Foundation (E.C.) and Umeå University (K.C., E.C.). J.A.D. is an Investigator and M.J. is a Research Specialist of the Howard Hughes Medical Institute. K.C. is a fellow of the Austrian Doctoral Program in RNA Biology and co-supervised by R. Schroeder. We are grateful to A. Witte, U. Bläsi and R. Schroeder for helpful discussions, financial support to K.C and hosting K.C. in their laboratories at MFPL. M.J., K.C., J.A.D. and E.C. have filed a related patent.
Footnotes
Supplementary Materials. The Supplementary Materials contain the Supplementary Materials and Methods, Supplementary Figures S1-S15 with legends, and Supplementary Tables S1-S3, with references 39–47.
References and Notes
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement
NIHMS995853-supplement-Supplement.pdf (6.2MB, pdf)
