Single cell sequencing for immunologists is revolutionizing our understanding of the immune system, enabling researchers to dissect cellular heterogeneity and identify novel immune cell populations. CONDUCT.EDU.VN offers a comprehensive resource to navigate the complexities of single cell RNA sequencing, providing immunologists with the knowledge to select the optimal method for their research. This guide explores various scRNA seq technologies, experimental applications, and unbiased bioinformatics methods to integrate diverse datasets. Discover more about cellular genomics, transcriptome profiling, and molecular identifiers at CONDUCT.EDU.VN.
1. Introduction to Single-Cell Sequencing in Immunology
The immune system, a complex network of cells, tissues, and organs, is critical for host defense and maintaining homeostasis, including tissue development and metabolism (Wynn et al., 2013; Zmora et al., 2017). Traditionally, microscopy and flow cytometry have classified immune cells based on surface markers. However, phenotypic markers alone cannot fully resolve all immune cell types, as many are expressed by multiple cell lineages or differentially regulated during inflammation (Merad et al., 2013; Becher et al., 2014; Guilliams et al., 2014). Gene expression studies on bulk populations of sorted immune cells have provided insights, but they fail to capture the variability between individual cells or account for contamination by unrelated cell types, masking significant heterogeneity (Jaitin et al., 2015). This is particularly important in temporally dynamic processes like cell differentiation.
Advances in next-generation sequencing have enabled interrogation of the immune system at the single-cell level. Single cell RNA sequencing (scRNA seq) is now a cornerstone in immunological studies, resolving cellular heterogeneity, defining cell development and differentiation, unraveling hematopoiesis, and understanding gene regulatory networks (Zheng et al., 2017; Villani et al., 2017; Bjorklund et al., 2016; See et al., 2017; Paul et al., 2015; Schlitzer et al., 2015; Mass et al., 2016; Dixit et al., 2016; Jaitin et al., 2016; Shalek et al., 2013). A static snapshot of single-cell transcriptomes offers a powerful view of differentiation and activation states.
The rapid development of low-input RNA-seq methods has led to a proliferation of scRNA-seq protocols, each with unique advantages and limitations. Selecting the most appropriate method for a specific research question can be challenging. This guide lists four commonly used scRNA seq methods and discusses their strengths and limitations in terms of workflow, sensitivity, data quality, and cost. We also demonstrate unbiased single cell identification and data integration from different scRNA-seq protocols.
2. Overview of Single-Cell RNA-Sequencing Technologies
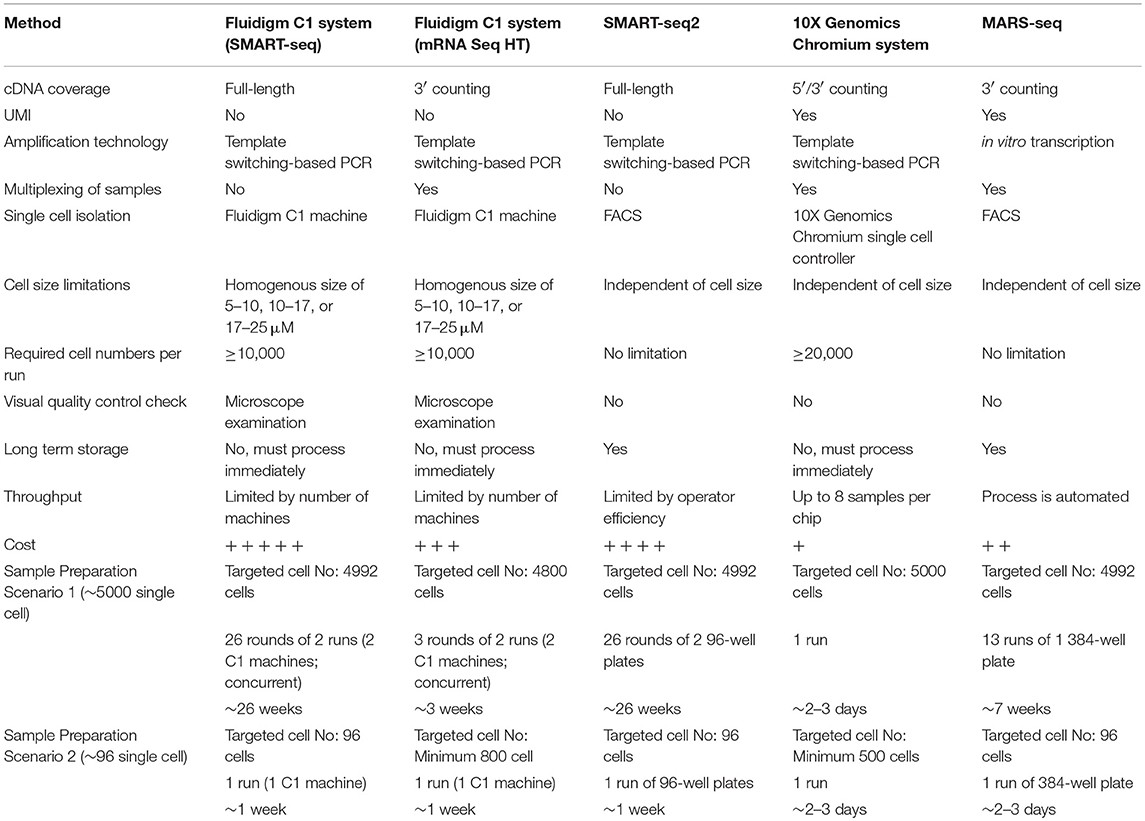
Since the first scRNA-seq protocol in 2009, numerous methods have emerged, differing in mRNA transcript amplification to generate full-length cDNA or cDNA with a unique molecular identifier (UMI). SMART-seq (switching mechanism at 5′ end of RNA template sequencing) and SMART-seq2 generate full-length cDNA, while MARS-seq (massively parallel RNA single-cell sequencing), STRT (single-cell tagged reverse transcription), CEL-seq (cell expression by linear amplification and sequencing), Drop-seq, and inDrops incorporate UMIs into cDNA (Tang et al., 2009; Ramskold et al., 2012; Picelli et al., 2013, 2014; Jaitin et al., 2014; Islam et al., 2011, 2014; Hashimshony et al., 2012, 2016; Macosko et al., 2015; Klein et al., 2015). Automation and sample preparation are facilitated by microfluidic or droplet-based platforms like Fluidigm C1, 10X Genomics Chromium, and InDrop from 1 CellBio. Alternative scRNA seq methods have been reviewed extensively (Giladi et al., 2018; Papalexi et al., 2018; Hedlund et al., 2018; Valihrach et al., 2018).
This review focuses on MARS-seq, SMART-seq2, Fluidigm C1, and 10X Genomics Chromium, widely used by biomedical scientists. These methods can be combined with fluorescence-activated cell sorting (FACS) to separate cells from heterogeneous suspensions. “Index sorting” using FACS isolates individual cells with known characteristics and records their location, allowing retrospective analysis. This approach avoids predefined cell sorting strategies, identifying better isolation strategies for downstream experiments. It also provides controls to determine which cell types are most sensitive to methodological biases.
2.1 Massively Parallel RNA Single Cell Sequencing (MARS-seq)
MARS-seq is an automated scRNA seq method where single cells are FACS-sorted into 384-well plates containing lysis buffer (Jaitin et al., 2014). The plates can be stored for long periods, offering flexibility. The method is unrestricted by cell size, shape, homogeneity, or total number. MARS-seq uses a 3′ end-counting mRNA sequencing method, generating partial cDNA transcripts tagged with barcodes and a unique molecular identifier (UMI) during reverse transcription, before pooling and amplification by in vitro transcription (IVT). The UMI enables quantitation of gene expression levels, reducing technical variability and bias (Kivioja et al., 2011; Grun et al., 2014). Pooling simplifies the process and increases throughput. This method detects approximately 500–3,000 genes per primary cell.
2.2 Fluidigm C1 Single Cell Full Length Messenger RNA (mRNA) Sequencing
The Fluidigm C1 is an automated microfluidic system that captures and processes up to 96 individual cells for mRNA quantitation. Cell capture, lysis, reverse transcription, and cell multiplexing occur in an integrated fluidic circuit (IFC) chip. Three cell size cartridges (5–10, 10–17, and 17–25 μM) are available, but input cells must be of uniform size and shape. A minimum of 10,000 cells is required, making it unsuitable for rare populations. Cells must be fresh and processed immediately, complicating integration with long experiments. Each machine accommodates only one cartridge at a time, requiring multiple machines for concurrent runs. The high cost of microfluidic cartridges can limit sample size. The C1 system allows individual visualization of captured cells, excluding empty wells, doublets, or debris. It employs SMART sequencing, generating full-length cDNA and detecting 300–7,000 genes per primary cell. The C1 mRNA Seq HT assay increases throughput to 800 cells, but uses 3′ end-counting mRNA sequencing, losing read coverage across the entire transcript.
2.3 Switching Mechanism at 5′ End of RNA Template (SMART-seq2)
SMART-seq2, an improved version of SMART-seq, refines reverse transcription, template switching, and pre-amplification to increase cDNA library yield and length (Picelli et al., 2014). SMART-seq2 generates full-length cDNAs, providing good read coverage for detecting gene isoforms or allele-specific expression using single-nucleotide polymorphisms (SNPs). However, it cannot incorporate UMIs or barcodes, complicating gene level quantification or multiplexing. Like MARS-seq, cells are sorted into 96- or 384-well PCR plates pre-filled with lysis buffer, compatible with index sorting. SMART-seq2 is not restricted by cell size, shape, homogeneity, or numbers. Reactions occur in individual wells requiring manual pipetting, making it time-consuming and increasing variability, but liquid handling robots can mitigate this. This method detects a higher number of genes per primary cell (~4,000–7,000).
2.4 10X Genomics Chromium Single Cell RNA Sequencing
The 10X Genomics Chromium system encapsulates single cells in gel beads in emulsion (GEMs). Each gel bead is labeled with oligonucleotides consisting of a unique barcode, a 10 bp UMI, sequencing adapters/primers, and an anchored 30 bp oligo-dT (Zheng et al., 2017). This system offers high throughput and reduces the need for sorting equipment. Up to eight samples can be processed simultaneously. Downstream processing (reverse transcription, cDNA amplification, and library construction) is simple, with reactions performed together in a single tube. This platform detects 500–1,500 genes per primary cell. While cost-effective and time-saving, this protocol offers little control over cell input and can be susceptible to biases. Rare cell populations may not be represented if insufficient cell numbers are analyzed, and users cannot determine which cells are collected prior to processing. The 10X Genomics Chromium system can be used with cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq), detecting multiplexed protein markers with unbiased transcriptome profiling (Stoeckius et al., 2017). Briefly, cells are stained with antibodies-oligo complexes prior to scRNA-seq. Stained cells are encapsulated, lysed, and antibody-derived oligos anneal via their 3′ poly A tails to gel beads, indexed by a shared cellular barcode during reverse transcription. CITE-seq can study post-translational gene regulation or large-scale immunophenotyping, enhancing discovery of cellular phenotypes, especially those with subtle transcriptomic differences.
3. Choosing the Right Platform: A Pragmatic Approach
Advancements in sequencing and computational methods are making scRNA-seq more accessible. Selecting the appropriate platform depends on the biological question, balanced against cell numbers, information depth, and cost. Estimating the expected level of cellular heterogeneity is crucial.
3.1 Which Protocol Should I Use?
The choice depends on the research question. The four approaches can be categorized into full-length methods (SMART-seq2 and Fluidigm C1) and molecular tag-based methods (MARS-seq and 10X Genomics Chromium). Full-length methods cover the entire transcriptome, increasing mappable reads, suitable for cell-type discovery, tissue composition assessment, allelic gene expression analysis, and isoform discovery. However, they cannot be multiplexed via sample pooling, increasing cost and labor, and UMIs cannot be incorporated.
Molecular tag-based methods sequence the 5′ or 3′ end of the molecule, combined with UMIs to enable multiplexing, improving gene expression quantification and throughput. Reads are restricted to one end of the transcript, reducing sensitivity. Despite this, the low cost and high throughput of tag-based approaches make them widely used for gene expression levels, cell-type discovery, and tissue composition studies.
Platform sensitivity determines sequencing depth and total genes detected per cell. Sensitivity is defined as the minimum input RNA molecules required for confident detection of a spike-in control, allowing detection of weakly expressed genes. Studies suggest that 1 million reads per cell is sufficient for saturated gene detection (Svensson et al., 2017; Ziegenhain et al., 2017). MARS-seq, Fluidigm C1, and SMART-seq2 detect a median of 4,763, 7,572, and 9,138 genes, respectively, consistent with our analysis of data from these platforms. SMART-seq2 outperforms other methods in sensitivity due to more mappable reads, as transcripts of tag-based methods may have proximal sequence features difficult to align to the genome.
3.2 How Many Cells Do I Need to Sequence?
The required number of cells depends on the research objective. Studies aiming to describe the immune landscape or discover rare cell populations can use a breadth-based approach, sequencing hundreds to tens of thousands of cells to provide a reasonable tissue composition distribution. This has been used to map multiple tissues including spleen, brain, and intestine (Jaitin et al., 2014; Zeisel et al., 2015; Rosenberg et al., 2018; Grun et al., 2015).
Amit and colleagues dissected cellular diversity within mouse spleen using MARS-seq, identifying eight transcriptionally distinct groups corresponding to B cells, natural killer cells, macrophages, monocytes, and four dendritic cell (DC) subpopulations (Jaitin et al., 2014). A separate study mapping murine brain heterogeneity sequenced 3,005 cells from mouse primary somatosensory cortex region S1 and hippocampal region CA1 using the Fluidigm C1 platform, identifying 47 molecularly distinct subclasses corresponding to known major cell types (Zeisel et al., 2015). These studies suggest that the required cell number depends on the number of discrete cellular states within the population. In a heterogenous population with transcriptionally distinct and equally distributed cellular states, 1,000–2,000 single cells could be sufficient for de novo clustering. If the cell of interest has a distinct transcriptional profile, it may be revealed with fewer cells and at a shallower sequencing depth. With the popularity of droplet-based technologies, there will be an increase of low sequencing depth studies examining 10- to 100-fold more cells (Zheng et al., 2017; Macosko et al., 2015; Klein et al., 2015). Researchers should consider which approach best suits their research questions and budget.
3.3 What Are Some Potential Applications of scRNA-seq?
scRNA-seq has numerous immunological applications. Traditionally, immune cells were considered homogenous, but scRNA-seq studies have revealed that populations can comprise transcriptionally distinct populations sharing overlapping phenotypic markers (Bjorklund et al., 2016; See et al., 2017; Gaublomme et al., 2015; Keren-Shaul et al., 2017). Bjorklund et al. identified four distinct innate lymphocyte cell (ILC) clusters in human tonsils corresponding to known ILC populations, namely ILC1-3 and natural killer (NK) cells, and uncovered three subpopulations within ILC3 (Bjorklund et al., 2016). Gury-BenAri et al. assessed the heterogeneity of helper-like ILC in the mouse small intestine, revealing 15 transcriptional states and functional plasticity within the subsets (Gury-BenAri et al., 2016). scRNA-seq can help reveal cellular heterogeneity masked in traditional phenotypic studies.
scRNA-seq can profile tissues and identify molecular drivers of disease. Gaublomme et al. profiled T helper 17 cells in experimental autoimmune encephalomyelitis in mice, showing high heterogeneity and transcriptional signatures correlated with pathogenicity (Gaublomme et al., 2015). Keren-Shaul et al. identified disease-associated microglia (DAM) interacting and phagocytizing plaques in Alzheimer’s disease (Keren-Shaul et al., 2017). Such studies can improve understanding of immune responses and pathogenicity, and pave the way for new therapeutic agents.
scRNA-seq can study immune function, such as antigen receptor repertories. T cell receptor sequences can be assembled from scRNA-seq reads and mapped against a reference pool (Stubbington et al., 2016). Stubbington et al. identified transcriptional states within a single expanded T cell clonotype during Salmonella infection in mice (Stubbington et al., 2016). A similar tool has been developed for B cell receptors (Canzar et al., 2017). These applications provide a better understanding of adaptive immunity responses to infection, autoantigens, or vaccination, and drive development in therapeutic approaches.
In a developmental context, Giladi et al. recently dissected hematopoietic stem cell differentiation trajectories in murine bone marrow, tracking their development into each hematopoietic lineage at single cell resolution, generating an unbiased reference model of hematopoiesis (Giladi et al., 2018). The global scientific community is collaborating to establish a “human cell atlas” using scRNA-seq technologies to map every cell type in the human body (Regev et al., 2017). When complete, this atlas will advance understanding of human physiology and impact biology and medicine.
4. Case Study: Resolving Dendritic Cell Ontogeny with scRNA-seq
Dendritic cells (DCs) are a cell type of interest, being small in numbers and heterogeneous in subsets (Dress et al., 2018). Human peripheral blood mononuclear cells consist of approximately 90% lymphocytes, 10% monocytes, and 1% dendritic cells. A recent report using the 10X Genomics Chromium system performed scRNA-seq on 68,000 unsorted peripheral blood mononuclear cells (PBMC) to identify various immune cell populations (Zheng et al., 2017). The study identified major immune cell populations, but found it difficult to identify or resolve cell types whose frequency was less than 1%. Enriching rare cell types in the sample prior to scRNA-seq, for example by pre-sorting using known surface markers, may be necessary.
Villani and colleagues focused on lineage−HLA-DR+ cells, comprising known blood DCs and monocytes, performing SMART-seq2 on 2,400 lineage−HLA-DR+ single cells, detecting transcriptionally distinct cell clusters identified using novel surface markers (Villani et al., 2017). This allowed their isolation by FACS and subsequent analysis by scRNA-seq to validate transcriptional identity, identifying new types of DCs and monocytes, as well as a novel DC precursor population. Our group focused on human blood lineage−HLA-DR+CD135+ cells, consisting of both DC subsets and their precursors, performing MARS-seq on 710 lineage−HLA-DR+CD135+ single cells (See et al., 2017). We identified two transcriptionally distinct clusters of plasmacytoid DC (pDC), two subpopulations of conventional DC (cDC), and a new cluster constituting pre-DC. Further interrogation of this novel pre-DC population revealed distinct lineage-committed sub-populations (one early “uncommitted” CD123high pre-DC subset, and two CD45RA+CD123low lineage-committed subsets with distinct functional features). These studies demonstrate that different scRNA seq platforms can be successfully applied to similar biological questions in complementary ways.
5. Computational Approach for Cell Type Identification of Unknown Single Cells
Before scRNA-seq, cell types were defined using antibodies against pre-selected cell surface markers. As technologies advanced, the number of markers per cell measured by flow cytometry or mass cytometry increased from <10 to >40. This allows detailed dissection of cellular heterogeneity, but lags behind the resolution possible with unbiased methods employing transcriptomic or proteomic techniques. scRNA-seq technologies can measure the transcriptomes of thousands of cells quickly, and computational methods have made robust cell identification possible. However, a challenge is knowing how to cluster the data and/or perform cell identification. Many algorithms are used to cluster single cell data, including shared nearest neighbor (SNN), SNN-Cliq, pcaReduce, clustering through imputation and dimensionality reduction (CIDR), single-cell consensus clustering (SC3), single cell RNA-seq profiling analysis (SINCERA), rare cell type identification (RaceID), GiniClust, and single-cell latent variable model (scLVM) (Waltman et al., 2013; Xu et al., 2015; Zurauskiene et al., 2016; Lin et al., 2017; Kiselev et al., 2017; Guo et al., 2015; Jiang et al., 2016; Buettner et al., 2015). After identifying cell clusters, differentially expressed genes in each cluster are determined and assigned as known/novel cell types.
Here, we explore the use of cell-type identification by estimating relative subsets of RNA transcripts (CIBERSORT) in unbiased cell type identification of single-cell transcriptomes, analyzing human peripheral blood mononuclear cells (PBMC) scRNA-seq data from two studies using either the 10X Genomics Chromium or SMART-seq2 platforms (Newman et al., 2015; Zheng et al., 2017; Villani et al., 2017). Zheng et al. performed single-cell RNA-seq of 68,000 human PBMC using the 10X Genomics Chromium system, clustering the cells into 10 subsets and identifying cluster-specific gene expression patterns (Zheng et al., 2017). Cell types in each cluster were inferred by aligning cluster-specific genes to known markers of PBMC populations, as well as comparing against the scRNA-seq profile of 11 purified PBMC subsets. Single-cell transcriptomes were compared with the average transcriptomes of the 11 purified populations by Spearman’s correlation. Each cell was assigned the same identity as the purified population with which it had the highest correlation, consistent with marker-based methods. For both analyses, cluster 9 contained monocytes and DC, whereas cluster 10 contained DC only. Cells from cluster 9 and 10 were extracted for further analysis and segregated into 4 sub-populations when further analyzed using the Seurat package (Satija et al., 2015). These 4 sub-clusters were visually verifiable on the t-Distributed Stochastic Neighbor Embedding (tSNE) reduced dimensions plots. tSNE visualizes high-dimensional similarities of cells in a two-dimensional map, plotting cells with similar properties close together, allowing interpretation of each cell type based on location (Maaten et al., 2008; Amir et al., 2013). Single cells were initially identified as different lineages via correlation with the purified PBMC populations superimposed on the tSNE plot. Cluster 2 comprised mainly DC, while clusters 0, 1 and 3 comprised mainly CD14+ monocytes. Although correlation-based cell type classification is largely consistent with clustering methods, some cells located in monocyte clusters were identified as DC.
To resolve whether these cells were indeed monocytes or true DC, we performed cell type identification via CIBERSORT analysis using the monocyte and DC gene signatures defined by the single-cell transcriptomic data (i.e., from the 11 purified PBMC subsets). First, we extracted CD14+ monocytes and DC and calculated the average gene expression level per cell type. Genes with maximum expression >0.0001 UMI were selected for CIBERSORT analysis, and percentage enrichment of signature genes was calculated for each individual cell, allowing assignment of lineage identity according to the most highly enriched gene sets. When this CIBERSORT-based cell type classification was overlaid on the tSNE plot, we observed much higher concordance with both clustering and tSNE segregation.
In this study with the 10X Genomics Chromium system, reference populations of purified PBMC allowed classification of unsorted single cell transcriptomes into 11 major immune cell types. We combined DC from cluster 9 and 10 and then further grouped these into distinct subsets. DCs in human blood are known to comprise two populations of cDC (CD141+ cDC1 and CD1c+ cDC2) as well as a subset of pDC, and a distinct population of pre-DCs. In our study (See et al., 2017), we sorted pure populations of CD141+ cDC1, CD1c+ cDC2, pDCs, and pre-DCs from human blood and generated bulk microarray data, from which we derived characteristic gene signatures for each subset. We next validated these gene signatures against published SMART-seq2 single cell data for each of the four DC populations described in Villani et al. Single cells pooled from each population were subjected to tSNE dimension reduction and then clustered into four subsets using the Seurat package. CIBERSORT comparison of these data against microarray-derived gene signatures allowed computational inference of cellular identities that were highly concordant with classification by FACS. Sorted populations of CD141+ cDC1, CD1c+ cDC2, and pDC were largely assigned to the corresponding cell type by CIBERSORT, with only a small portion of each being classified as pre-DC (likely representing progenitor cells committed to cDC1 or cDC2 fates, as well as uncommitted pre-DC that share phenotypic similarities with pDC). More intriguingly, the majority of sorted double negative cells were predicted to be pre-DCs, suggesting that this compartment may contain genuine cDC precursors. It was not possible to identify some cell types where permutation p-values were >0.05. However, despite the fact that cell sorting for microarray and SMART-seq2 was performed by two independent labs, this work confirmed that the signatures derived from microarray were able to aid lineage identification of SMART-seq2 single cells via CIBERSORT. We therefore proceeded to apply the same gene signatures to the prediction of cell types for single cells analyzed with the 10X Genomics Chromium dataset. We first performed tSNE dimension reduction and clustering of individual DC using Seurat, and overlaid CIBERSORT-inferred cell types on the tSNE plot. Among the 3 clusters generated, cluster 2 comprised predominantly of CD141+ cDC1. Unsupervised clustering was in line with cell type inference using sorted cells, suggesting that the conventional marker-based identification of cDC1 is well-defined and can be validated using a marker-free approach. In contrast, cluster 0 represented a mixed population of CD1c+ and undetermined cells, whereas cluster 1 comprised a mixture of pDC, pre-DC and undetermined cells. These findings are consistent with earlier reports that CD1c+ cDC2 in fact represent a heterogeneous population of poorly characterized composition, whereas pDCs are phenotypically similar to pre-DC. Cluster 0 and cluster 1 were assigned as CD1c+ cDC2, and pDC, respectively. Compared with SMART-seq2, the 10X Genomics Chromium dataset generated a higher number of unsorted cells that were labeled as undetermined. These cells were not significantly enriched in signatures of cDC1, cDC2, pDC, or pre-DC, suggesting that these could be unknown subsets acquired by marker-free scRNA-seq of unsorted cells.
In summary, we used two different methods, Spearman’s correlation and CIBERSORT, to identify cell types in the 10X Genomics Chromium PBMC dataset. We found that CIBERSORT performed slightly better than did a correlation-based approach. A major reason for this could be that CIBERSORT first identified signature genes for each cell type, followed by an additional step of vector regression to calculate a gene signature enrichment score. In any case, both methods use bulk transcriptomes for reference and are thus highly dependent on the cell types present in the reference dataset. Accordingly, the use of a comprehensive dataset that is directly relevant to the study of interest will significantly improve the accuracy of cell type identification.
6. Data Integration and Correction of Technical Variation
With the increased data yield provided by scRNA-seq, researchers can now mine existing datasets to perform multiple different types of analysis. Datasets generated by different scRNA seq platforms often require integration prior to downstream analysis, and technical variation between datasets must be corrected before these can be combined. When applying scRNA-seq to a large number of cells, experiments are usually carried out in batches, resulting in inter-assay variability that can conceal biological heterogeneity.
Villani et al. performed SMART-seq2 on two separate batches of sorted cDC1, cDC2, double negative DC, and pDC, before performing t-SNE dimension reduction and clustering analysis, which identified two distinct sub-populations for each input cell type (Villani et al., 2017). Overlaying batch information onto the tSNE plot revealed that these sub-populations corresponded to the two separate assay runs. To remove this batch effect, the Seurat package implements the canonical correlation analysis (CCA) algorithm, which identifies the dimensions in which batch 1 and 2 have the highest correlation and projects the cells onto these dimensions. After CCA normalization, the same cell types from batch 1 and batch 2 were well-aligned, with no evident separation of cells between assay runs.
Next, we used CCA to integrate single-cell data as generated by SMART-seq2 method and 10X Genomics Chromium system. Single cells isolated from purified cDC1, cDC2, double negative cells, and pDC populations were prepared using SMART-seq2. Single cell data from unsorted PBMCs were generated by 10X Genomics Chromium system and only DC were isolated for integration with SMART-seq2 data. DCs from the 10X Genomics Chromium experiment were inferred based on CIBERSORT analysis as mentioned previously. Before CCA normalization, cells from SMART-seq2 method and 10X Genomics Chromium system were well-separated, and two distinct subsets were identified for each lineage, reflecting the use of the two different analytical platforms. After CCA normalization, the cells analyzed by each platform became well-mixed and were clustered mainly by cell type. Notably, the double negative cells that were previously separated from other lineages were also observed to merge with the CD1c+ population after CCA. We next attempted to integrate datasets of slightly different cellular composition by adding monocytes to the 10X Genomics Chromium data, whereas the SMART-seq2 dataset still comprised DC only. Cells were clustered mainly by cell type regardless of the platform used, except that double-negative DCs were now allocated to the monocyte cluster and some CD141+ cells were now present in the CD1c+ cluster.
Our analysis indicates that CCA is able to correct batch effect confounders when no other biological factors differ between experimental replicates. The CCA algorithm make the assumption that data from both batches have the same or similar cellular composition. It is important to note that CCA can still force batches to align even if they have dissimilar cellular composition, which can result in masking of genuine biological variation. To overcome this limitation, the mutual nearest neighbor (MNN) algorithm can be employed to identify similar cell populations or “pairs” that are present in both batches (Haghverdi et al., 2018). MNN pairs are used to calculate analytical drift between assay runs and subsequently compensate batch effect for all cells present. In the absence of any shared structure, a cell population of known composition (e.g., a cell line) can also be spiked into each sample in order to remove batch effects by providing a uniform reference population. While both CCA and MNN are powerful tools, several other normalization techniques (both current and future) may further improve batch effect correction in the years ahead. However, a thorough comparison of these novel methods will be required using variable input data in order to identify which approaches best suit which datasets.
7. Conclusion: The Future of Single-Cell Sequencing in Immunology
In this review, we discussed the strengths and limitations of widely used scRNA seq platforms, as well as technical barriers to analyzing single-cell transcriptome datasets. As next generation sequencing techniques and computational methods continue to improve, scRNA-seq in immunological studies will become more widespread and routine. Once a complete set of reference databases or “immune mapping” studies has been completed, new strategies will be required to multiplex single-cell profiling with other techniques that permit analysis of multiple molecular features of individual cells in parallel.
As the complexity of these technologies increases, investigator choice of analytical platform must be carefully guided by specific hypotheses and biological questions, hopefully leading to deeper insight into the role of the immune system in health and disease.
Navigating the complexities of single-cell sequencing can be challenging, but CONDUCT.EDU.VN is here to help. Our comprehensive resources provide the guidance and support you need to succeed in your research. Visit CONDUCT.EDU.VN today to explore our articles, tutorials, and expert advice.
Address: 100 Ethics Plaza, Guideline City, CA 90210, United States
Whatsapp: +1 (707) 555-1234
Website: conduct.edu.vn
8. Frequently Asked Questions (FAQ) about Single Cell Sequencing
1. What is single-cell RNA sequencing (scRNA-seq)?
Single-cell RNA sequencing (scRNA-seq) is a high-throughput technique used to examine the gene expression profiles of individual cells within a population. It provides insights into cellular heterogeneity, gene regulation, and cell differentiation.
2. Why is scRNA-seq important in immunology?
In immunology, scRNA-seq is crucial for understanding the complexity of the immune system. It helps in identifying new immune cell types, studying immune responses, and understanding the molecular drivers of diseases.
3. What are the main steps involved in a scRNA-seq experiment?
The main steps include cell isolation, cell lysis, reverse transcription, cDNA amplification, library construction, sequencing, and data analysis.
4. What are some common scRNA-seq methods/platforms?
Common methods/platforms include MARS-seq, SMART-seq2, Fluidigm C1, and 10X Genomics Chromium.
5. How do I choose the right scRNA-seq method for my research?
The choice depends on the research question, balanced against factors like cell numbers, information depth, cost, and whether you need full-length transcripts or tag-based methods.
6. What is the role of Unique Molecular Identifiers (UMIs) in scRNA-seq?
UMIs are used to label individual RNA molecules, which helps in reducing amplification bias and improving the accuracy of gene expression quantification.
7. How many cells do I need to sequence in a scRNA-seq experiment?
The number of cells depends on the research objective. For discovering rare cell populations, sequencing hundreds to tens of thousands of cells may be necessary.
8. What are some common computational tools for analyzing scRNA-seq data?
Common tools include Seurat, CIBERSORT, and algorithms for clustering like SNN, pcaReduce, and CIDR.
9. What is batch effect correction, and why is it important?
Batch effect correction is a process used to remove technical variation between different batches of scRNA-seq data. It is important to ensure that biological variation is not masked by technical artifacts.
10. How can I integrate data from different scRNA-seq platforms?
Data can be integrated using methods like canonical correlation analysis (CCA) and mutual nearest neighbor (MNN) algorithms to correct technical variations between datasets.

